Highlights
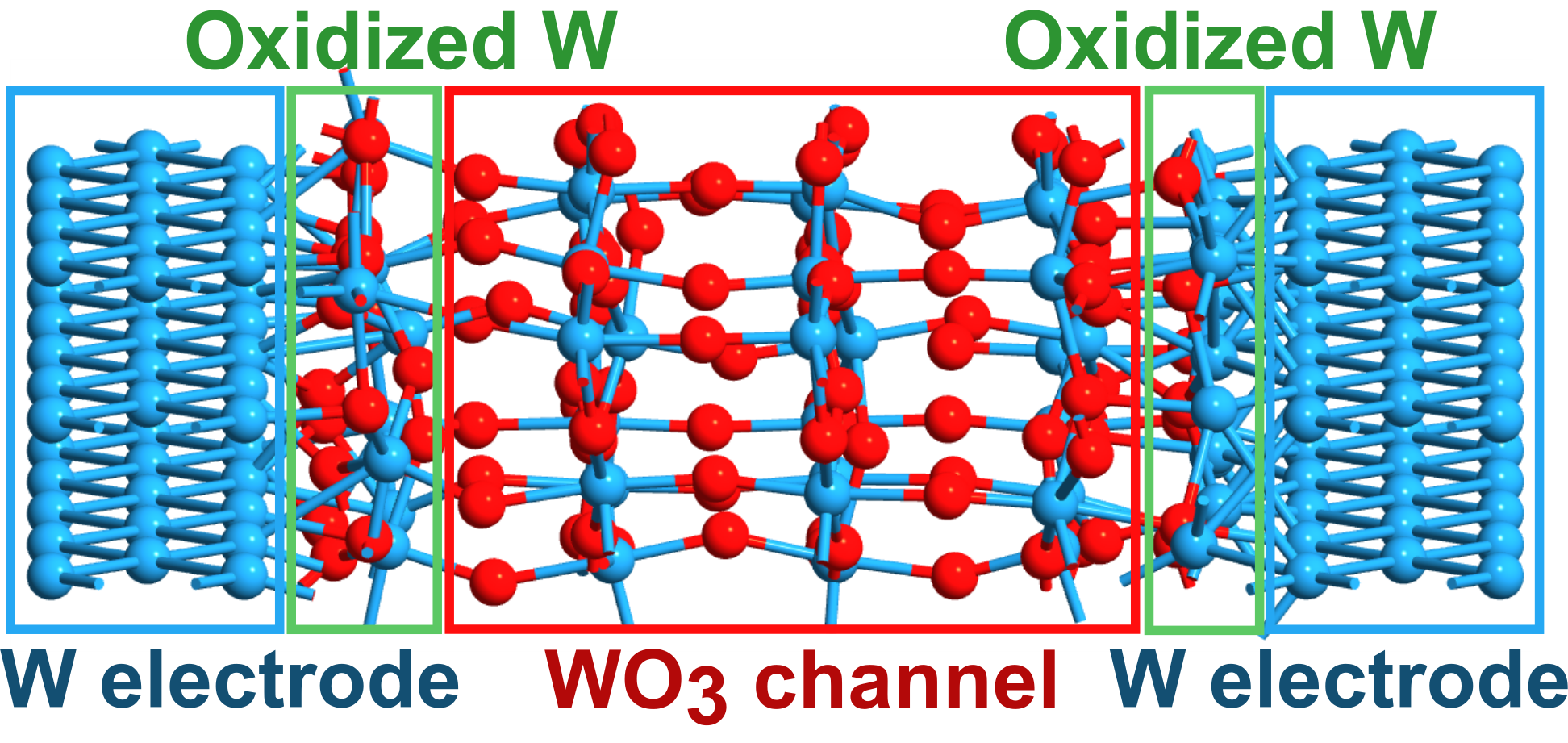
Nanoscale Electron Transport through WO3 and HxWO3
Tungsten trioxide (WO3) is a cornerstone material for emerging oxide nanoelectronics, yet its transport behavior at the ultimate scaling limits remains poorly understood. In this work, we employ first-principles quantum transport simulations, combining density functional theory (DFT) with the non-equilibrium Green’s function (NEGF) formalism, to investigate the electronic properties of HxWO3 junctions in the sub 5-nm regime. We identify a critical structural scaling threshold of six molecular layers (~26.9 Å), below which quantum tunneling dominates, and above which stable semiconducting behavior and effective conductance modulation emerge. Using this 6-layer framework, we demonstrate that hydrogen intercalation WO3 in induces a monotonic enhancement in current density, providing a 2.5-fold increase in conductance through the filling of tungsten 5d states. Furthermore, our analysis reveals that nanoscale transport is highly sensitive to the spatial configuration of protons; specifically, OHO defects at bridging oxygen sites induce significant structural distortions (~ 178° to 163°) that attenuate current by reducing orbital overlap. These results establish the fundamental scaling limits for WO3 integration and provide a microscopic origin for the structural sensitivity of protonic transport, offering a predictive framework for the design of high-density neuromorphic and gas-sensing architectures.

First-Principles Insights into Dopant-Vacancy Interactions in Acceptor-Doped Perovskite Oxides: A Comparative Study of A- and B-Site Substitution
Understanding and controlling dopant-vacancy interactions is central to the design of high-performance oxide-ion conductors. Using density functional theory, we systematically examined oxygen vacancy stability in acceptor-doped perovskite oxides (LaAlO₃, LaGaO₃, and SrTiO₃) with substitution at both A and B sites. Enumeration of all symmetry-inequivalent vacancy configurations reveals that, in the absence of first-nearest-neighbor dopants, vacancy energies scale linearly with a simple Coulombic metric defined by dopant-vacancy distances, consistent with electrostatic attraction. Importantly, configurations in which dopants occupy first-nearest-neighbor sites deviate strongly from this electrostatic trend with notably different behavior for A site and B site dopants. Analysis of local structure and bonding uncovers the origin of those deviations from the electrostatic trends. The proximity of an A site dopants could suppress the characteristic umbrella-like lattice relaxation of the BO6 octahedra that stabilizes isolated vacancies, reducing any elastic component. Concurrently, harder ions on dopant sites lead to loss of partial covalency in adjacent cation-oxygen bonds destabilizing near-neighbor dopant-vacancy configurations. These coupled electrostatic, elastic, and bonding effects govern site-dependent dopant-vacancy interactions and establish a unified physical framework for minimizing vacancy trapping in perovskite oxides.
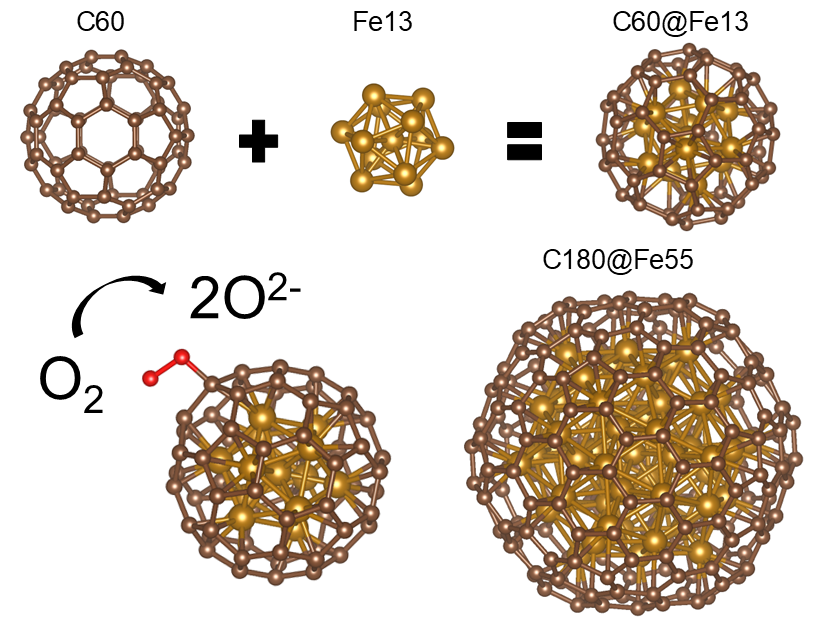
Oxygen Activation on Carbon-coated Iron Nanoparticles
This work explores the feasibility of molecular oxygen activation and dissociation on the sp²-hybridized carbon surface of carbon-coated iron nanoparticles. Using density functional theory with a generalized gradient approximation, we elucidate the geometry and electronic structure of these nanoparticles, highlighting the nature of the C–Fe binding interactions and the resulting modifications to the carbon surface electronic states. The enhanced catalytic activity of carbon induced by the underlying iron core is attributed to core–shell electronic interactions within the nanoparticles. Activation of molecular oxygen to superoxo and peroxo species was investigated using the nudged elastic band method, with electron transfer processes analyzed in detail and linked to the core–shell characteristics of the system. Additionally, we examined the effects of nitrogen doping in the carbon shell on the structural and electronic properties of the nanoparticles. Potential degradation pathways, including parasitic reactions during oxygen activation, were also identified. This study offers new theoretical insights into the functional behavior of Fe–C–N catalysts.
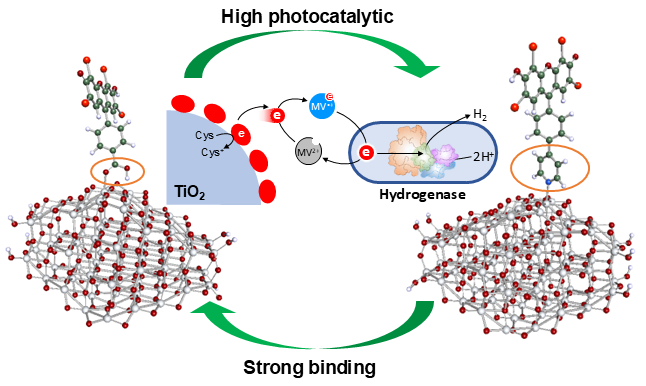
Improved Charge Transfer Performance of Eosin Y-Sensitized Anatase TiO2 by Anchoring Group Modification: from the Theoretical Design to Experiment
Theoretical design and experimental proof of photocatalytic performance of Eosin Y (EY) on anatase TiO2 with pyridine linker were performed for increasing photobiocatalytic activity of water splitting using visible light. Comparative studies on the hybrid interface of anatase and EY with the carboxyl and pyridine anchor were performed by density functional theory (DFT), time-dependent density functional theory (TD-DFT) calculations and experimental photoreduction of methyl viologen (MV). The geometries, binding interactions between dyes and anatase, electronic structures and electron transfer as well as the effect of isomers (ortho, meta, para) on the dye / anatase systems were investigated. Theoretical results indicated that EY with carboxyl and pyridine anchors had visible adsorption and electron transfer from the dye to the anatase titania. Compared to carboxyl-para, which had the best optical performance among carboxyl groups, the adsorption strength of pyridine-ortho was close to that of carboxyl-para, while the oscillator strength increased significantly, which was more than 10 times higher than that of carboxyl-para. Corresponding with the theoretical estimation, EY pyridine linked TiO2 is active to MV reduction under visible light irradiation by fast charge transfer, in particular, pyridine-ortho and para. Furthermore, high stability is also achieved on pyridine-para. Apparent quantum yield higher than 2.00% and 0.67 % at 520 nm light was experimentally achieved on biocatalytic H2 and NH3 formation, respectively, on EY pyridine linked TiO2, which was correctly predicted by the DFT calculations.
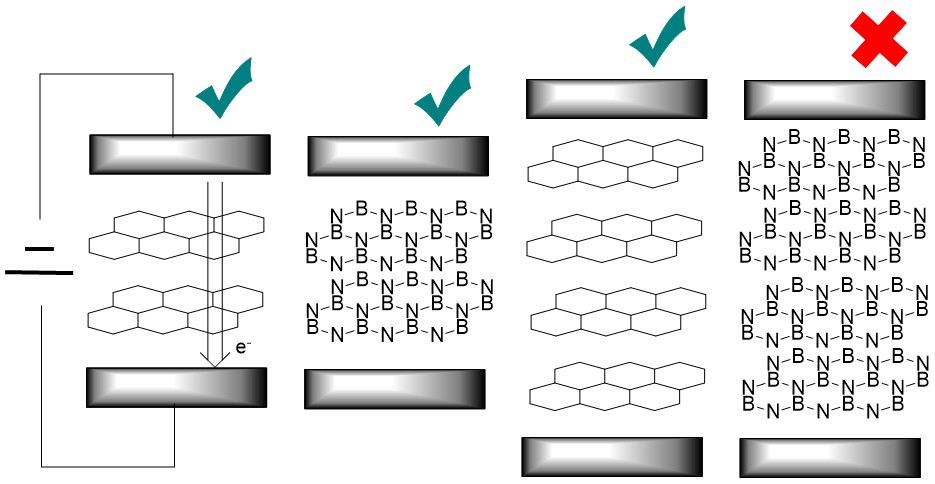
Electron Transport through Multilayer Nanographene and Hexagonal Boron Nitride Junctions
In this study, we employ the Non-Equilibrium Green’s Function (NEGF) method combined with density functional theory (DFT) to compare electron transport through several layers of nanographene and hexagonal boron nitride (h-BN). Calculations were performed for 2, 4, and 6 layers, corresponding to thicknesses of 1 nm, 2 nm, and 3 nm, respectively. Electron transport was computed perpendicular to the layers in the stacking direction. We compared the decay of the current with the number of layers and evaluated the ability of h-BN to filter currents as a material coating. To investigate the effect of disorder, we included two major defects in the graphene lattice: nitrogen doping and Stone-Wales defects. Nitrogen doping transforms graphene from a zero-band gap semiconductor to metal, while Stone-Wales defects open the band gap. For h-BN, we considered Stone-Wales defects. A detailed comparison of electron transport through five materials: multilayer nanographene, N-doped multilayer nanographene, Stone-Wales defective multilayer nanographene, h-BN, and Stone-Wales defective h-BN; allowed us to understand the currents at the nanoscale and the chemical and structural control over the electron transport. The slopes of the current decay with thickness enabled us to extrapolate trends for electron transport in thicker multilayer carbon and h-BN materials.
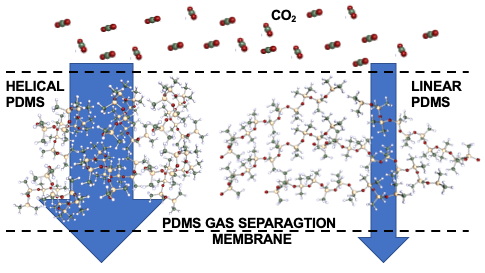
Curvature Effect in Polydimethylsiloxane Interaction with CO2
In this study we employ density functional theory to investigate the binding interaction between polydimethylsiloxane and CO2 for application in gas separation membranes. The binding strength has been studied systematically as a function of the monomer conformational rotations in the polymer chain. Our work identified major differences between the CO2 interaction with the helical conformation and linear conformation of polydimethylsiloxane polymer chains. We have further estimated dependence between the CO2 binding strength and the polydimethylsiloxane polymer chain curvature by systematically evaluating the CO2 binding to cyclic polydimethylsiloxane oligomers. The enhanced CO2 interaction with helical chains and cyclic oligomers was attributed to cooperative, confinement effects and local electron density distribution at the Si-O-Si fragments. The binding modes were identified using vibration frequency analysis.